基因富集分析,是分析基因表达信息的一种方法。
富集是将基因按照先验知识,即基因组注释信息,对基因进行分类的过程。能够帮助寻找到的基因是否存在共性。
涵盖了基因的
- 细胞组分(cellular component), 指基因产物位于何种细胞器或基因产物组中(如糙面内质网,核糖体,蛋白酶体等),即基因产物在什么地方起作用。
- 分子功能(molecular function), 描述在个体分子生物学上的活性,如催化活性或结合活性。
- 生物学过程(), 由分子功能有序地组成的,具有多个步骤的一个过程。
系统分析基因功能与基因组信息的数据库,它整合了基因组学、生物化学和系统功能组学的信息.
主要的特点是将基因与各种生化反应联系在了一起。提供了整合代谢途径查询。
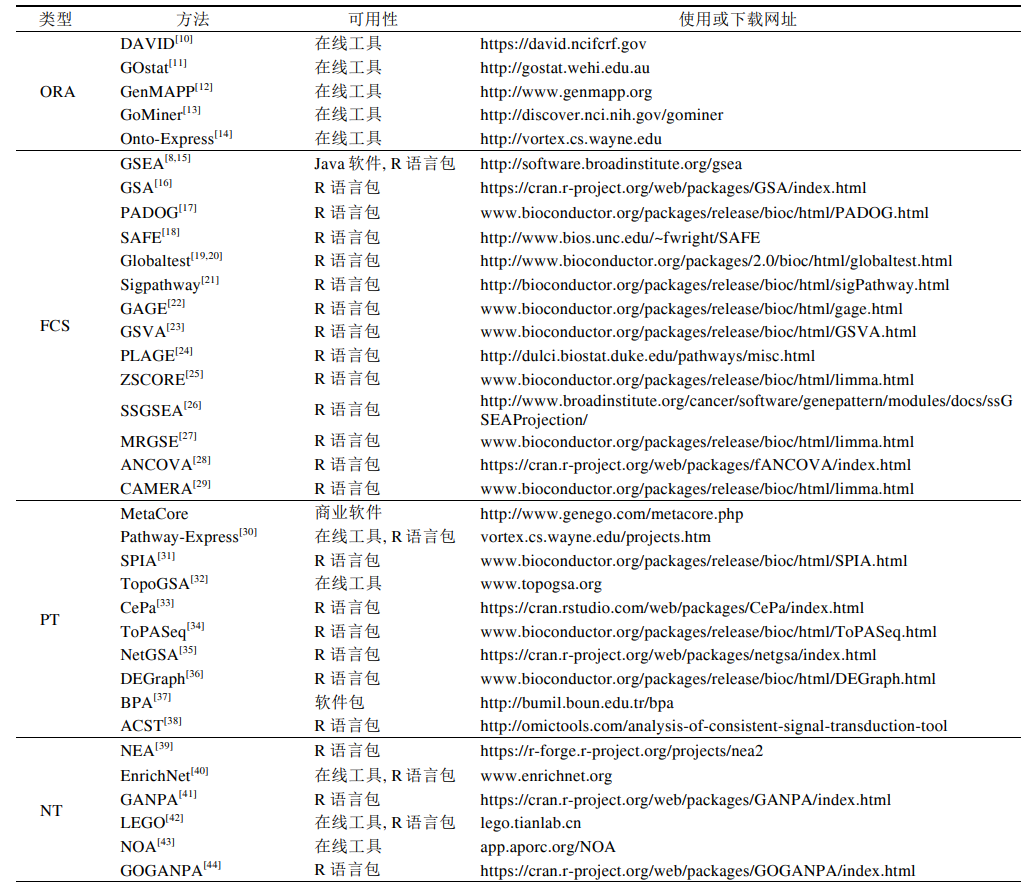
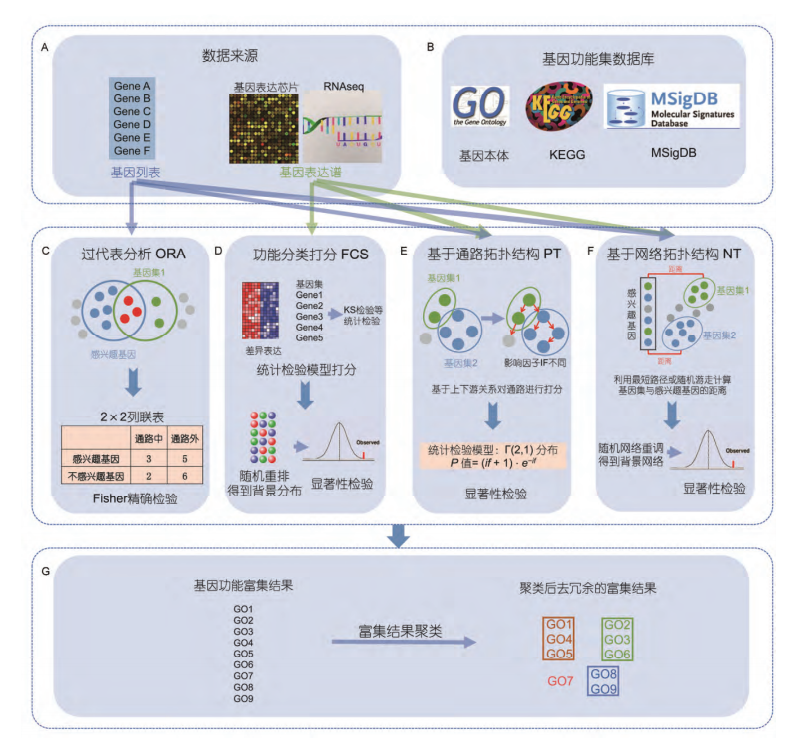
基于数据来源和算法可将富集分析方法分为四大类:
- ORA
- FCS
- PT
- NT
-
获得一组感兴趣的基因(一般是差异表达基因)
-
与基因集做交集,找出共同的基因并计数
-
检验
差异基因 其他基因 在基因集中 10 50 不在基因集中 290 19950
- 仅使用了基因数目信息
- 假设每个基因都是独立的
The hypothesis of functional class scoring (FCS) is that although large changes in individual genes can have significant effects on pathways, weaker but coordinated changes in sets of functionally related genes can also have significant effects
- 对基因组中所有基因表达水平的差异值打分或者排序
- 计算富集分数
- 利用随机抽样检验统计值得显著水平
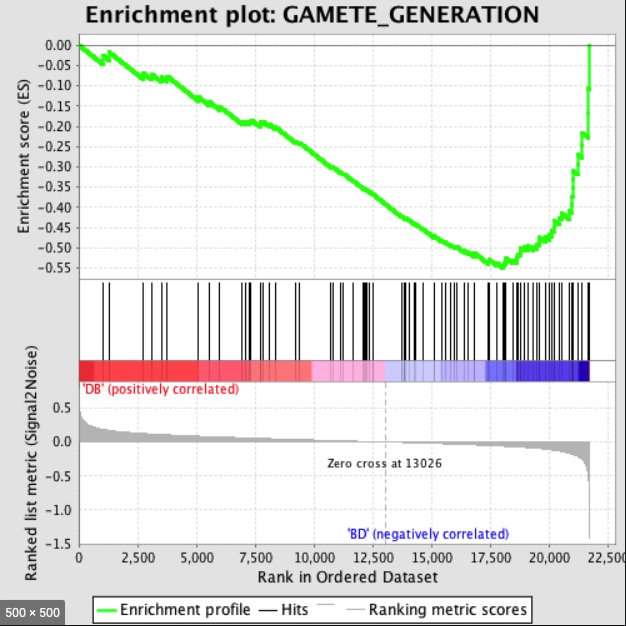
给定一个排序的基因表L和一个预先定义的基因集S ,GSEA的目的是判断S里面的成员s在L里面是随机分布还是主要聚集在L的顶部或底部。若研究的基因集S的成员显著聚集在L的顶部或底部,则说明此基因集成员对表型的差异有贡献。
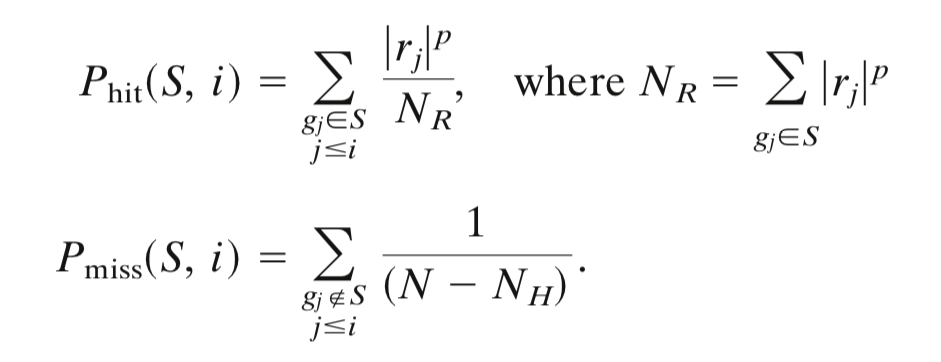
The ES is the maximum deviation from zero of
$P_{hit} - P_{miss}$
ORA 和 FCS 方法在进行通路的富集分析时, 都将通路中的每个基因视作独立个体, 而实际上通路内的基因存在调控、被调控、相互作用等复杂的. 因而, 在进行通路的富集分析时, 需要考虑到通路中基因的生物学属性. 例如, 在一个调控通路中, 上游基因的表达水平改变显然要远大于下游基因的表达水平改变对整个通路的影响. 基于通路拓 扑结构的 PT 富集分析方法就是把基因在通路中的位置(上下游关系), 与其他基因的连接度和调控作用类型等信息综合在一起来评估每个基因对通路的贡献并给予相应的权重, 然后再把基因的权重整合入功能富集分析.
利用数据库中的基因相互作用关系来间接地把基因的生物学属性整合入富集分析.
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("clusterProfiler")data(geneList, package = "DOSE")
gene <- names(geneList)[abs(geneList) > 2]
gene.df <- bitr(gene, fromType = "ENTREZID",
toType = c("ENSEMBL",
"SYMBOL"),
OrgDb = org.Hs.eg.db) # Genome wide annotation for Human
head(gene.df) ENTREZID ENSEMBL SYMBOL
1 4312 ENSG00000196611 MMP1
2 8318 ENSG00000093009 CDC45
3 10874 ENSG00000109255 NMU
4 55143 ENSG00000134690 CDCA8
5 55388 ENSG00000065328 MCM10
6 991 ENSG00000117399 CDC20go <- enrichGO(gene = gene,
universe = names(geneList),
OrgDb = org.Hs.eg.db,
ont = "CC"
readable = T)
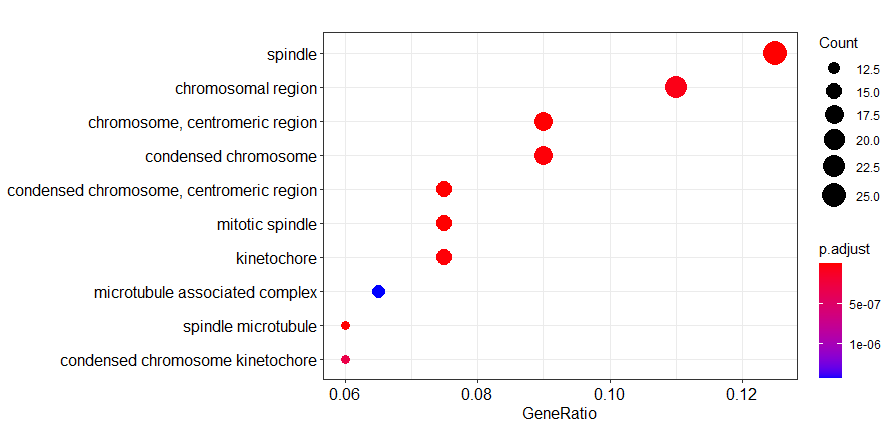
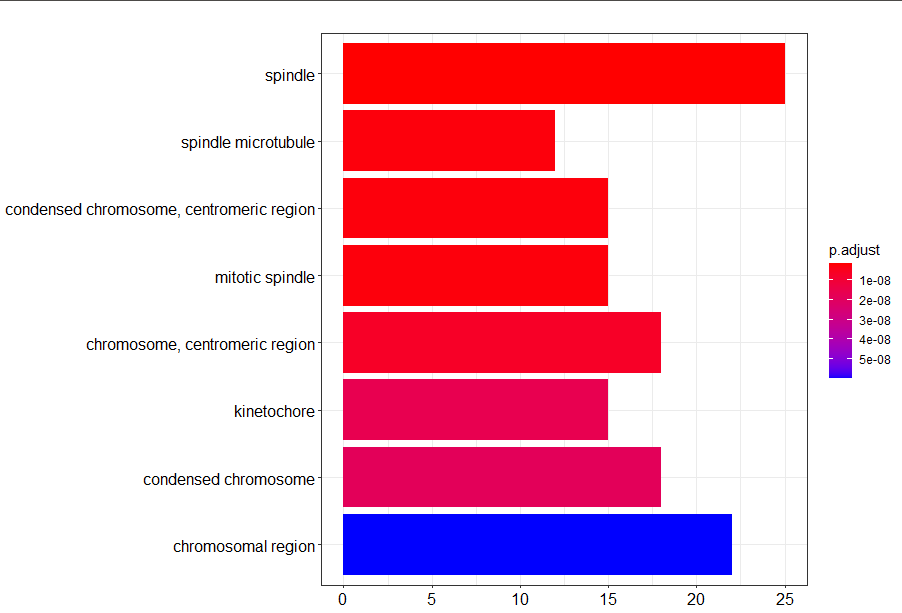
head(go) ID Description GeneRatio BgRatio
GO:0005819 GO:0005819 spindle 25/200 272/11816
GO:0005876 GO:0005876 spindle microtubule 12/200 48/11816
GO:0000779 GO:0000779 condensed chromosome, centromeric region 15/200 91/11816
GO:0072686 GO:0072686 mitotic spindle 15/200 92/11816
GO:0000775 GO:0000775 chromosome, centromeric region 18/200 154/11816
GO:0000776 GO:0000776 kinetochore 15/200 107/11816
pvalue p.adjust qvalue
GO:0005819 4.695505e-12 1.493171e-09 1.378996e-09
GO:0005876 1.623758e-11 2.581776e-09 2.384361e-09
GO:0000779 2.808998e-11 2.628919e-09 2.427899e-09
GO:0072686 3.306816e-11 2.628919e-09 2.427899e-09
GO:0000775 1.061302e-10 6.749884e-09 6.233756e-09
GO:0000776 3.047081e-10 1.614953e-08 1.491466e-08 dotplot(go2)
barplot(go2)
go3 <- gseGO(geneList = geneList,
OrgDb = org.Hs.eg.db,
ont = "CC",
nPerm = 1000,
minGSSize = 100,
maxGSSize = 500,
pvalueCutoff = 0.05,
verbose = FALSE)head(ego3) ID
GO:0031012 GO:0031012
GO:0099568 GO:0099568
GO:0062023 GO:0062023
GO:0044441 GO:0044441
GO:0005788 GO:0005788
GO:0032838 GO:0032838
Description
GO:0031012 extracellular matrix
GO:0099568 cytoplasmic region
GO:0062023 collagen-containing extracellular matrix
GO:0044441 ciliary part
GO:0005788 endoplasmic reticulum lumen
GO:0032838 plasma membrane bounded cell projection cytoplasm
setSize enrichmentScore NES
GO:0031012 427 -0.4868578 -2.144854
GO:0099568 368 -0.3426037 -1.484557
GO:0062023 342 -0.5200592 -2.241048
GO:0044441 298 -0.3612491 -1.544017
GO:0005788 257 -0.4102832 -1.733604
GO:0032838 137 -0.4365603 -1.710997
pvalue p.adjust qvalues rank
GO:0031012 0.001256281 0.0358209 0.0258108 1797
GO:0099568 0.001308901 0.0358209 0.0258108 2613
GO:0062023 0.001331558 0.0358209 0.0258108 1897
GO:0044441 0.001340483 0.0358209 0.0258108 2643
GO:0005788 0.001369863 0.0358209 0.0258108 1836
GO:0032838 0.001466276 0.0358209 0.0258108 2417
leading_edge
GO:0031012 tags=37%, list=14%, signal=33%
GO:0099568 tags=28%, list=21%, signal=23%
GO:0062023 tags=39%, list=15%, signal=34%
GO:0044441 tags=32%, list=21%, signal=25%
GO:0005788 tags=27%, list=15%, signal=23%
GO:0032838 tags=28%, list=19%, signal=23% gseaplot(go3, geneSetID = "GO:0031012")
主要是将上述函数enrichGO等替换为KEGG
kegg <- enrichKEGG(gene = gene,
organism = "hsa",
pvalueCutoff = 0.05)
head(kegg)dotplot(kegg)
barplot(kegg)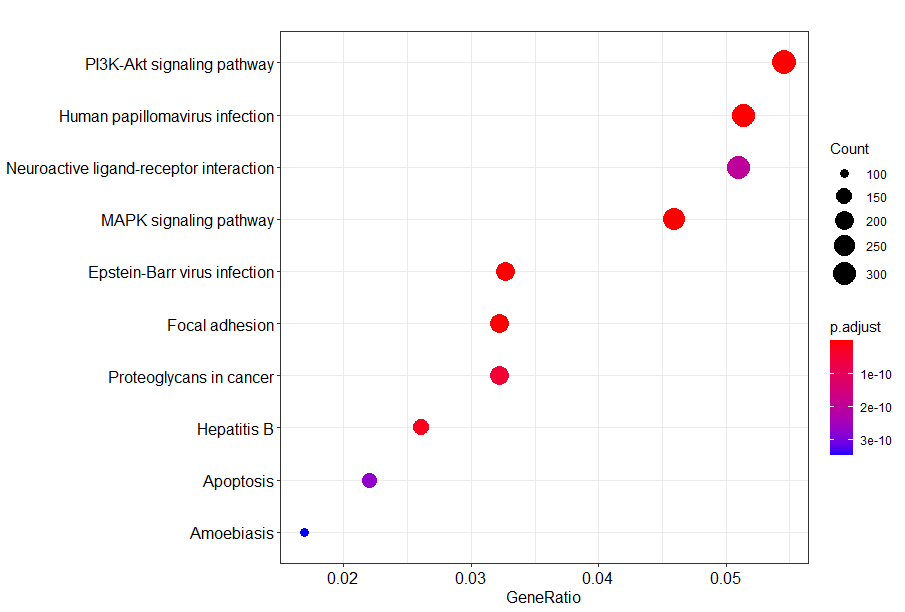

kegg2 <- gseKEGG(geneList = geneList,
organism = 'hsa',
nPerm = 1000,
minGSSize = 120,
pvalueCutoff = 0.05,
verbose = FALSE)
gseaplot(kegg2, geneSetID = "hsa04151")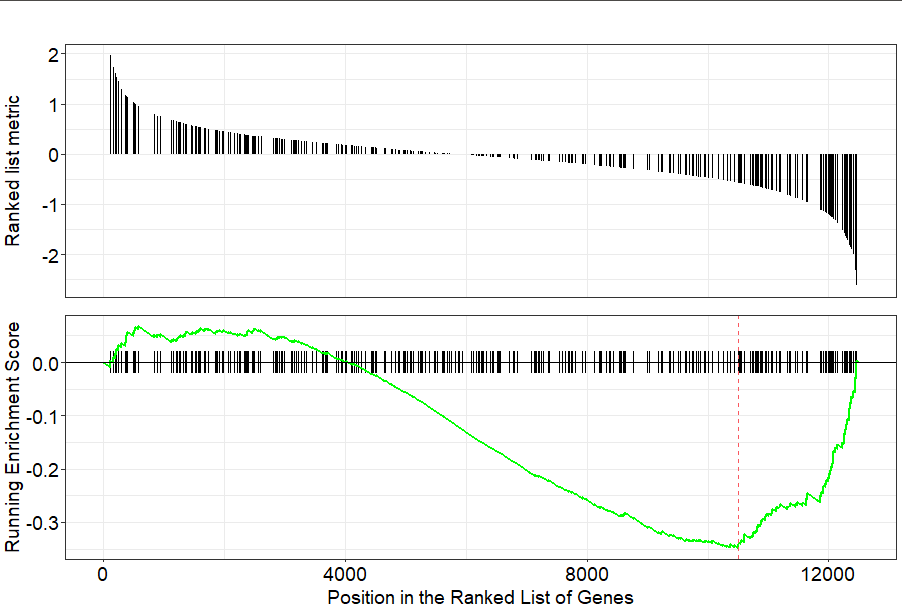
https://yulab-smu.github.io/clusterProfiler-book/chapter12.html
- Khatri, P., Sirota, M. & Butte, A. J. Ten Years of Pathway Analysis: Current Approaches and Outstanding Challenges. PLOS Computational Biology 8, e1002375 (2012).
- Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences 102, 15545–15550 (2005).
- Ma, J., Shojaie, A. & Michailidis, G. A comparative study of topology-based pathway enrichment analysis methods. BMC Bioinformatics 20, 546 (2019). https://doi.org/10.1186/s12859-019-3146-1
- G Yu, LG Wang, Y Han, QY He. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS: A Journal of Integrative Biology 2012, 16(5):284-287. doi:10.1089/omi.2011.0118
- https://zhuanlan.zhihu.com/p/61511124
- https://www.plob.org/article/11508.html
- https://www.jianshu.com/p/47b5ea646932
- https://yulab-smu.github.io/clusterProfiler-book/
- 基因功能富集分析的研究进展