Reads crystallographic cif files, calculates crystal properties and simulates diffraction.
Version 3.3

{kind=link}
By Dan Porter, Diamond Light Source 2024
$ ipython -i -m Dans_Diffraction
OR
$ ipython -m Dans_Diffraction gui
"""Python Sctipt"""
import Dans_Diffraction as dif
xtl = dif.Crystal('some_file.cif')
print(xtl) # print Crystal structure parameters
# Print reflection list:
print(xtl.Scatter.print_all_reflections(energy_kev=5))
# Plot Powder pattern:
xtl.Plot.simulate_powder(energy_kev=8)
plt.show()
# Start graphical user interface:
xtl.start_gui()Full code documentation available here.
Try it out on mybinder!
For comments, queries or bugs - email [email protected]
Citation: If you use this code (great!), please cite the published DOI: 10.5281/zenodo.8106031
Requirements: Python 3+ with packages: Numpy, Matplotlib, Tkinter. BuiltIn packages used: sys, os, re, glob, warnings, json, itertools
Install stable version from PyPI:
$ python -m pip install Dans-Diffraction
Or, install the latest version direct from GitHub:
$ python -m pip install git+https://github.com/DanPorter/Dans_Diffraction.git
Or, Download the latest version from GitHub (with examples!):
$ git clone https://github.com/DanPorter/Dans_Diffraction.git
$ cd Dans_Diffraction
$ python -m pip install .
From version 3.2, installing Dans_Diffraction will include a run script for the gui:
$ dansdiffraction
Alternatively, Dans_Diffraction is best run within an interactive python environment:
$ ipython -i -m Dans_Diffraction
Dans_Diffraction can also be run in scripts as an import, example scripts are provided in the Examples folder.
import Dans_Diffraction as dif
xtl = dif.Crystal('some_file.cif')
xtl.info() # print Crystal structure parameters
help(xtl) # all functions (nearly!) are documentedxtl.Cell.latt([2.85,2.85,10.8,90,90,120]) # set lattice parameters
xtl.Atoms.info() # Print Symmetric positions
xtl.Structure.info() # Print All positions in P1 symmetry (same structure and functions as xtl.Atoms)
# Symmetric positions
xtl.Atoms.changeatom(idx=0, u=0, v=0, w=0, type='Co', label='Co1')
xtl.Atoms.addatom(idx=0, u=0, v=0, w=0, type='Co', label='Co1')
# After adding or changing an atom in the Atoms class, re-generate the full structure using symmetry arguments:
xtl.generate_lattice()
# Full atomic structure in P1 symmetry
xtl.Structure.changeatom(idx=0, u=0, v=0, w=0, type='Co', label='Co1')
xtl.Structure.addatom(idx=0, u=0, v=0, w=0, type='Co', label='Co1')
# Plot crystal Structure
xtl.Plot.plot_crystal() # 3D plot
xtl.Plot.plot_layers() # 2D plot for layered materials
xtl.Symmetry.info() # print symmetry arguments
xtl.Symmetry.addsym('x,y,z+1/2') # adds single symmetry operation
xtl.Symmetry.changesym(0, 'x,y,z+1/4')
xtl.Symmetry.load_spacegroup(194) # replaces current symmetry operations
# After adding or changing symmetry operations, regengerate the symmetry matrices
xtl.Symmetry.generate_matrices()Lattice parameters, crystal structure and symmetry operations will be saved to the CIF. If magnetic moments are defined, magnetic symmetry operations and moments will also be saved and format changed to "*.mcif".
xtl.write_cif('edited file.cif')X-ray or neutron structure factors/ intensities are calculated based on the full unit cell structure, including atomic form-factors (x-rays) or coherent scattering lengths (neutrons).
# Choose scattering options (see help(xtl.Scatter.setup_scatter))
xtl.Scatter.setup_scatter(scattering_type='x-ray', energy_keV=8.0)
# Allowed radiation types:
# 'xray','neutron','xray magnetic','neutron magnetic','xray resonant'
xtl.Scatter.print_all_reflections() # Returns formated string of all allowed reflections
inten = xtl.Scatter.intensity([h,k,l]) # Returns intensity
twotheta, iten, reflections = xtl.Scatter.powder(units='twotheta')
# Plot Experimental Intensities
xtl.Plot.simulate_powder() # Powder pattern
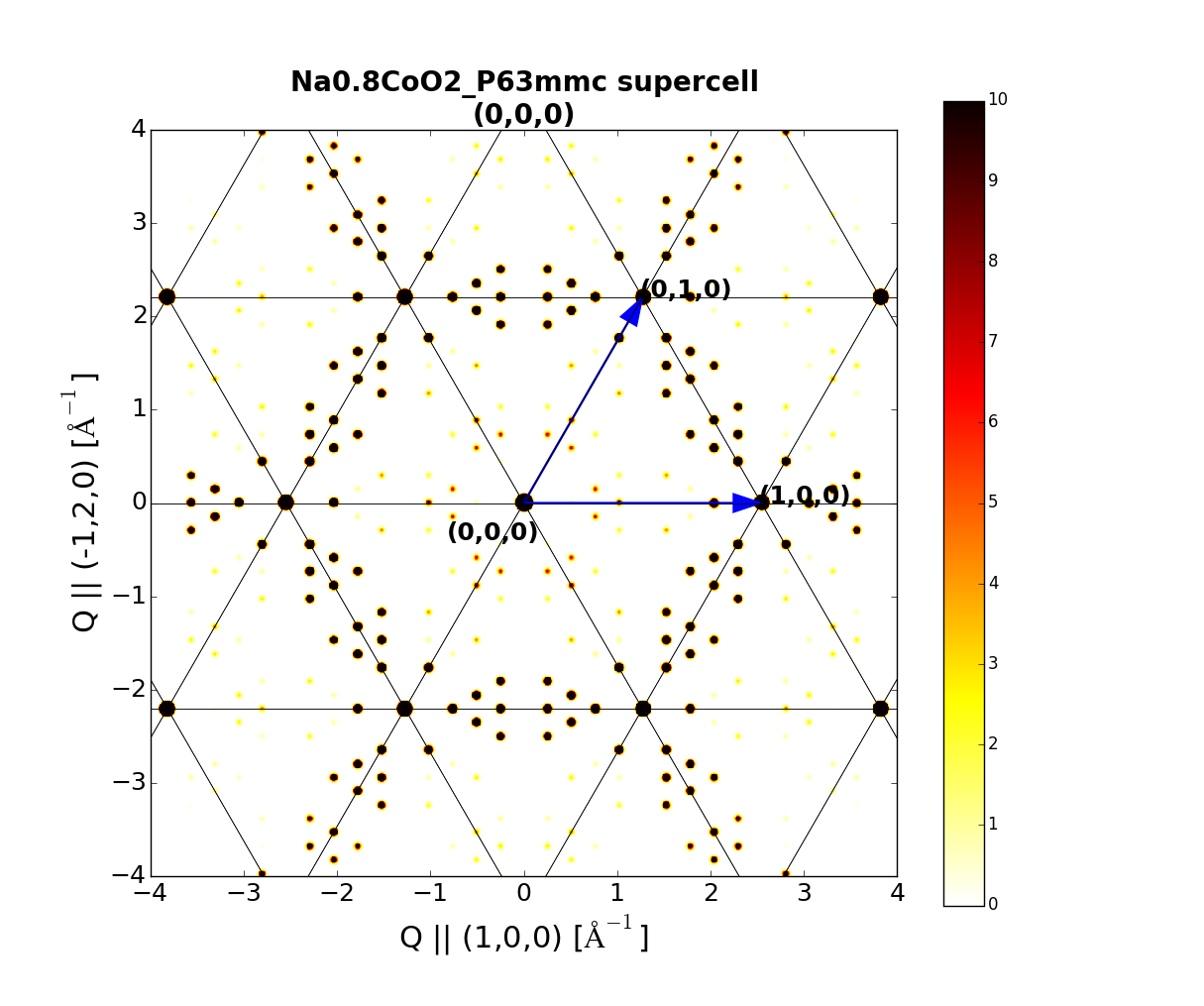
xtl.Plot.simulate_hk0() # Reciprocal space plane
Magnetic structures and scattering are currently in development and shouldn't be treated as accurate!
Simple magnetic structures can be loaded from magnetic cif (*.mcif) files. Magnetic moments are stored for each atomic position as a vector. The crystal object has a seperate set of magnetic symmetry operations. Symmetry operations from the tables of magnetic spacegroups can also be loaded. Only simple magnetic structures are allowed. There must be the same number of magnetic symmetry operations as crystal symmetry operations and atomic positions can only have single moments assigned.
xtl = dif.Crystal('some_file.mcif')
xtl.Atoms.mxmymz() # return magnetic moment vectors on each ion
xtl.Symmetry.symmetry_operations_magnetic # magnetic symmetry operations (list of strings)
xtl.Symmetry.print_magnetic_spacegroups() # return str of available magnetic spacegroups, given crystal's spacegroup
xtl.Symmetry.load_magnetic_spacegroup(mag_spg_number) # loads mag. operations given mag. spacegroup numberMagnetic scattering is also available for neutrons and x-rays (both resonant and non-resonant), using the appropriate magnetic form-factors.
Imag = xtl.Scatter.magnetic_neutron(HKL=[0,0,3])
Ires = xtl.Scatter.xray_resonant_magnetic(HKL=[0,0,3], energy_kev=2.838, azim_zero=[1, 0, 0], psi=0, polarisation='s-p', F0=0, F1=1, F2=0)Superstructures can be built using the Superstructure class, requring only a matrix to define the new phase:
su = xtl.generate_superstructure([[2,0,0],[0,2,0],[0,0,1]])Superstucture classes behave like Crystal classes, but have an additional 'Parent' property that references the original crystal structure and additional behaviours partiular to superstructures. Superstructures loose their parent crystal and magnetic symmetry, always being defined in P1 symmetry. So su.Atoms == su.Structure.
print(su.parent.info()) # Parent structure
su.P # superstructure matrix
su.superhkl2parent([h, k, l]) # index superstructure hkl with parent cell
su.parenthkl2super([h, k, l]) # index parent hkl with supercellScattering from different crystal structures can be compared using the MultiCrystal class:
xtls = xtl1 + xtl2
xtls.simulate_powder()The Crystal class contains a lot of atomic properties that can be exposed in the Properties class:
xtl.Properties.info()Calculated properties include:
- Molecular weight
- Density
- Diamagnetic suscpetibility
- x-ray absorption coefficient, attenuation length, transmission and refractive index
- Molecular charge balance
- Molecular mass fraction
- Atomic orbitals
- Magnetic exchange paths (in progress...)
Properties are calulated using the atomic structure along with atomic data stored in the folder Dans_Diffraction/data.
Simulations of multiple scattering at different azimuths for a particular energy can be simulated. Based on code by Dr Gareth Nisbet.
.
azimuth, intensity = xtl.Scatter.ms_azimuth([h,k,l], energy_kev=8)

Start a new GUI, then select a cif file:
$ ipython -i -m Dans_Diffraction gui
Or start the GUI from within the interactive console:
dif.start_gui()Using an already generated crystal:
xtl.start_gui()
New in version 3.0.0. Simulate a generic detector situated around the crystal sample, with the ability to control detector location shape and size and lattice orientation.
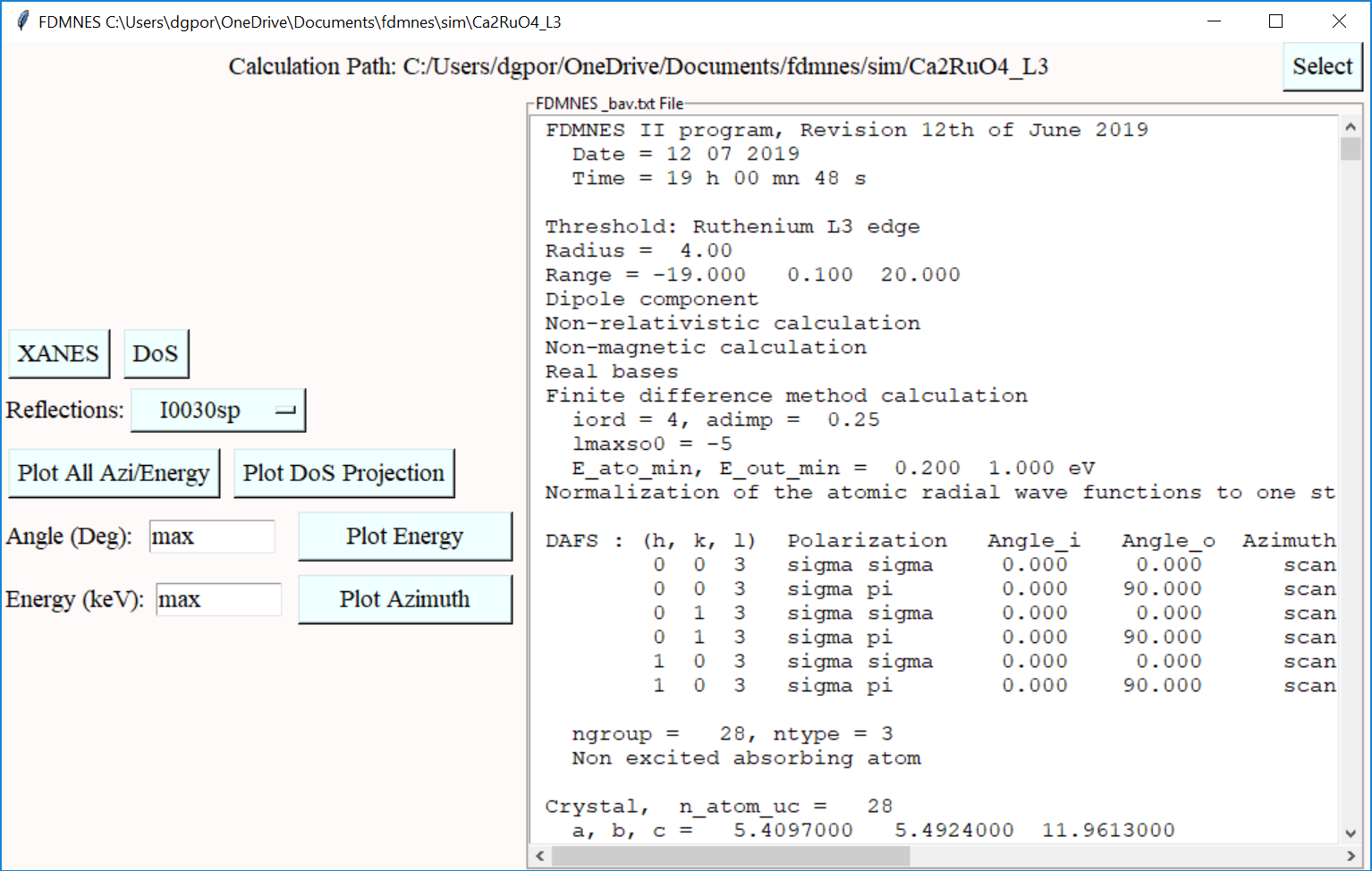
FDMNES is a powerful tool for simulating resonant x-ray diffraction, created by Y. Joly and O. Bunau.
The Dans_Diffraction FDMNES class allows for the automatic creation of input files and simple analysis of results. The following command should be used to activate these features (only needs to be issued once).
dif.activate_fdmnes()Once activated, the FDMNES classes become available.
fdm = dif.Fdmnes(xtl) # Create input files and run FDMNES
fdma = dif.FdmnesAnalysis(output_path, output_name) # Load output files and plot resultsSee class documentation for more information.
Once activated, FDMNES GUI elements become available from the main window, emulating functionality of the classes.

| Date | Thanks to... |
|---|---|
| 2018 | Thanks to Hepesu for help with Python3 support and ideas about breaking up calculations |
| Dec 2019 | Thanks to Gareth Nisbet for allowing me to inlude his multiple scattering siumulation |
| April 2020 | Thanks to ChunHai Wang for helpful suggestions in readcif! |
| May 2020 | Thanks to AndreEbel for helpful suggestions on citations |
| Dec 2020 | Thanks to Chris Drozdowski for suggestions about reflection families |
| Jan 2021 | Thanks to aslarsen for suggestions about outputting the structure factor |
| April 2021 | Thanks to Trygve Ræder for suggestions about x-ray scattering factors |
| Feb 2022 | Thanks to Mirko for pointing out the error in two-theta values in Scatter.powder |
| March 2022 | Thanks to yevgenyr for suggesting new peak profiles in Scatter.powder |
| Jan 2023 | Thanks to Anuradha Vibhakar for pointing out the error in f0 + if'-if'' |
| Jan 2023 | Thanks to Andreas Rosnes for testing the installation in jupyterlab |
| May 2023 | Thanks to Carmelo Prestipino for adding electron scattering factors |
| June 2023 | Thanks to Sergio I. Rincon for pointing out the rounding error in Scatter.powder |
| July 2023 | Thanks to asteppke for suggested update to Arrow3D for matplotlib V>3.4 |
| July 2023 | Thanks to Yves Joly for helpful suggestions on FDMNES wrapper |
| Jan 2024 | Thanks to Carmelo Prestipino for adding search_distance and plot_distance |
| April 2024 | Thanks to Innbig for pointing out an issue with liquid crystal simulations |
| May 2024 | Thanks to paul-cares pointing out a silly spelling error in the title! |
Copyright 2024 Diamond Light Source Ltd.
Licensed under the Apache License, Version 2.0 (the "License"); you may not use this file except in compliance with the License. You may obtain a copy of the License at
http://www.apache.org/licenses/LICENSE-2.0
Unless required by applicable law or agreed to in writing, software distributed under the License is distributed on an "AS IS" BASIS, WITHOUT WARRANTIES OR CONDITIONS OF ANY KIND, either express or implied. See the License for the specific language governing permissions and limitations under the License.
Files in this package covered by this licence:
- classes_crystal.py
- classes_scattering.py
- classes_plotting.py
- classes_properties.py
- classes_multicrystal.py
- classes_orientation.py
- classes_orbitals.py
- functions_general.py
- functions_plotting.py
- functions_scattering.py
- functions_crystallography.py
- tkgui/*.py
Other files are either covered by their own licence or not licenced for other use.
| Dr Daniel G Porter | [email protected] |
|---|---|
| www.diamond.ac.uk | Diamond Light Source, Chilton, Didcot, Oxon, OX11 0DE, U.K. |