![]()

![]()


Pasmopy is a scalable toolkit to identify prognostic factors for cancers based on intracellular signaling dynamics generated from personalized kinetic models. It is compatible with biomass and offers the following features:
- Construction of mechanistic models from text
- Personalization of the model using transcriptome data
- Prediction of patient outcome based on in silico signaling dynamics
- Sensitivity analysis for prediction of potential drug targets
Online documentation is available at https://pasmopy.readthedocs.io.
You can also build the documentation locally by running the following commands:
$ cd docs
$ make htmlThe site will live in _build/html/index.html.
The latest stable release (and required dependencies) can be installed from PyPI:
$ pip install pasmopy
Pasmopy requires Python 3.8+ to run.
This example shows you how to build a simple Michaelis-Menten two-step enzyme catalysis model with Pasmopy.
E + S ⇄ ES → E + P
An enzyme, E, binding to a substrate, S, to form a complex, ES, which in turn releases a product, P, regenerating the original enzyme.
import os
from pasmopy import Text2Model, create_model, run_simulation
# Prepare a text file describing the biochemical reactions (e.g., `michaelis_menten.txt`)
reactions = """\
E + S ⇄ ES | kf=0.003, kr=0.001 | E=100, S=50
ES → E + P | kf=0.002
"""
observables = """
@obs Substrate: u[S]
@obs E_free: u[E]
@obs E_total: u[E] + u[ES]
@obs Product: u[P]
@obs Complex: u[ES]
"""
simulation_condition = """
@sim tspan: [0, 100]
"""
with open("michaelis_menten.txt", mode="w") as f:
f.writelines(reactions)
f.writelines(observables)
f.writelines(simulation_condition)
# Convert the text into an executable model
description = Text2Model("michaelis_menten.txt")
description.convert()
assert os.path.isdir("michaelis_menten")
# Run simulation
model = create_model("michaelis_menten")
run_simulation(model)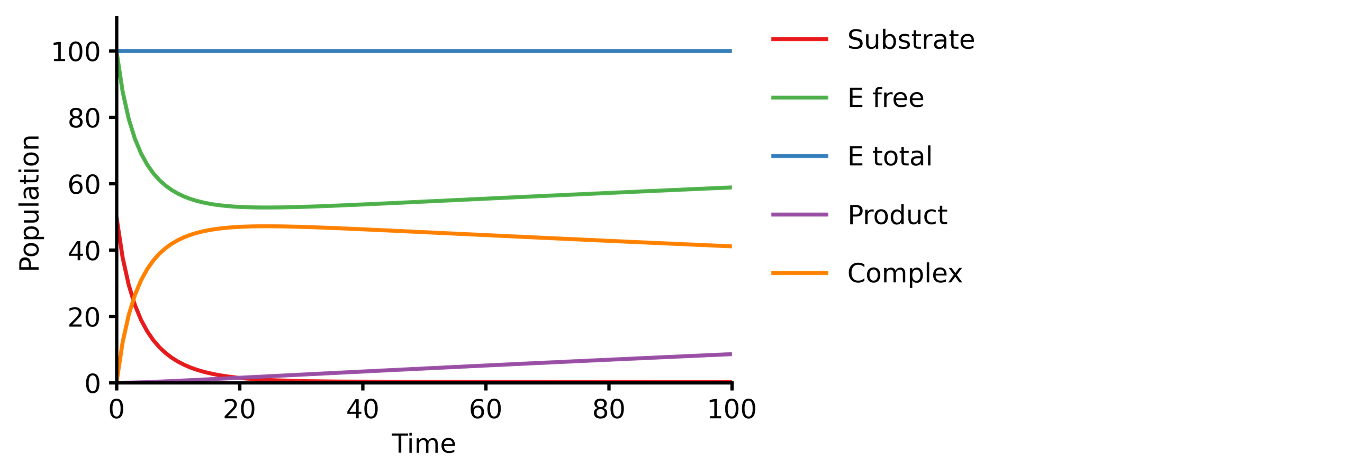
For more examples, please refer to the Documentation.
Using Pasmopy, we built a mechanistic model of ErbB receptor signaling network, trained with protein quantification data obtained from cultured cell lines, and performed in silico simulation of the pathway activities on breast cancer patients using The Cancer Genome Atlas (TCGA) transcriptome datasets. The temporal activation dynamics of Akt, extracellular signal-regulated kinase (ERK), and c-Myc in each patient were able to accurately predict the difference in prognosis and sensitivity to kinase inhibitors in triple-negative breast cancer (TNBC).
For further details, please see https://pasmopy.readthedocs.io/en/latest/personalized_model.html.
-
Imoto, H., Yamashiro, S. & Okada, M. A text-based computational framework for patient -specific modeling for classification of cancers. iScience 25, 103944 (2022). https://doi.org/10.1016/j.isci.2022.103944
-
Imoto, H., Yamashiro, S., Murakami, K. & Okada, M. Protocol for stratification of triple-negative breast cancer patients using in silico signaling dynamics. STAR Protocols 3, 101619 (2022). https://doi.org/10.1016/j.xpro.2022.101619